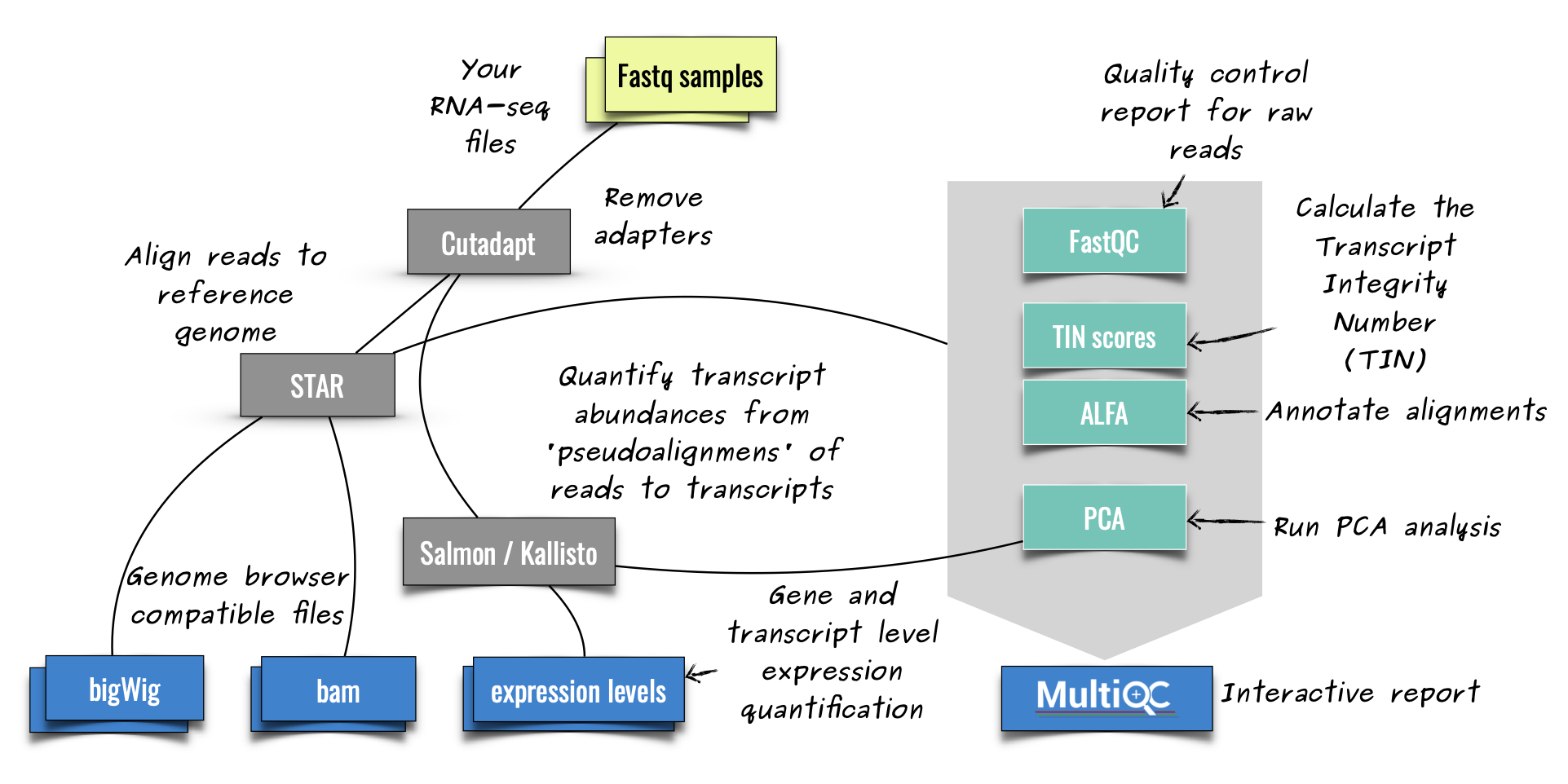
[Snakemake][snakemake] workflow that covers common steps of short read RNA-Seq
**ZARP** ([Zavolan-Lab][zavolan-lab] Automated RNA-Seq Pipeline) is a generic RNA-Seq analysis workflow that allows
library analysis developed by the [Zavolan lab][zavolan-lab].
users to process and analyze Illumina short-read sequencing libraries with minimum effort. The workflow relies on
publicly available bioinformatics tools and currently handles single or paired-end stranded bulk RNA-seq data.
The workflow is developed in [Snakemake][snakemake], a widely used workflow management system in the bioinformatics
community.
Reads are analyzed (pre-processed, aligned, quantified) with state-of-the-art
According to the current ZARP implementation, reads are analyzed (pre-processed, aligned, quantified) with state-of-the-art
tools to give meaningful initial insights into the quality and composition
tools to give meaningful initial insights into the quality and composition of an RNA-Seq library, reducing hands-on time for bioinformaticians and giving experimentalists the possibility to rapidly assess their data. Additional reports summarise the results of the individual steps and provide useful visualisations.
of an RNA-Seq library, reducing hands-on time for bioinformaticians and giving
experimentalists the possibility to rapidly assess their data.
Below is a schematic representation of the individual steps of the workflow
<divalign="center">
("pe" refers to "paired-end"):
<imgwidth="60%"src=images/zarp_schema.png>
</div>
> ![rule_graph][rule-graph]
For a more detailed description of each step, please refer to the [workflow
> **Note:** For a more detailed description of each step, please refer to the [workflow
documentation][pipeline-documentation].
> documentation][pipeline-documentation].
## Requirements
Currently the workflow is only available for Linux distributions. It was tested
# Requirements
on the following distributions:
The workflow has been tested on:
- CentOS 7.5
- CentOS 7.5
- Debian 10
- Debian 10
- Ubuntu 16.04, 18.04
- Ubuntu 16.04, 18.04
## Installation
> **NOTE:**
> Currently, we only support **Linux** execution.
### Cloning the repository
Traverse to the desired directory/folder on your file system, then clone/get the
# Installation
## 1. Clone the repository
Go to the desired directory/folder on your file system, then clone/get the
repository and move into the respective directory with:
repository and move into the respective directory with:
Workflow dependencies can be conveniently installed with the [Conda][conda]
Workflow dependencies can be conveniently installed with the [Conda][conda]
package manager. We recommend that you install
package manager. We recommend that you install[Miniconda][miniconda-installation]
[Miniconda][miniconda-installation]for your system (Linux). Be sure to select
for your system (Linux). Be sure to select Python 3 option.
Python 3 option. The workflow was built and tested with `miniconda 4.7.12`.
The workflow was built and tested with `miniconda 4.7.12`.
Other versions are not guaranteed to work as expected.
Other versions are not guaranteed to work as expected.
### Installing dependencies
## 3. Dependencies installation
For improved reproducibility and reusability of the workflow,
For improved reproducibility and reusability of the workflow,
each individual step of the workflow runs either in its own [Singularity][singularity]
each individual step of the workflow runs either in its own [Singularity][singularity]
container or in its own [Conda][conda] virtual environemnt. As a consequence, running this workflow has very few individual dependencies. However, for the **container execution** it requires Singularity to be installed on the system where the workflow is executed. As the functional installation of Singularity requires root privileges, and Conda currently only provides Singularity for Linux architectures, the installation instructions are
container or in its own [Conda][conda] virtual environemnt.
slightly different depending on your system/setup:
As a consequence, running this workflow has very few individual dependencies.
The **container execution** requires Singularity to be installed on the system where the workflow is executed.
As the functional installation of Singularity requires root privileges, and Conda currently only provides Singularity
for Linux architectures, the installation instructions are slightly different depending on your system/setup:
#### For most users
### For most users
If you do *not* have root privileges on the machine you want to run the
If you do *not* have root privileges on the machine you want
workflow on *or* if you do not have a Linux machine, please [install
to run the workflow on *or* if you do not have a Linux machine, please [install
Singularity][singularity-install] separately and in privileged mode, depending
Singularity][singularity-install] separately and in privileged mode, depending
on your system. You may have to ask an authorized person (e.g., a systems
on your system. You may have to ask an authorized person (e.g., a systems
administrator) to do that. This will almost certainly be required if you want
administrator) to do that. This will almost certainly be required if you want
to run the workflow on a high-performance computing (HPC) cluster. We have
to run the workflow on a high-performance computing (HPC) cluster.
successfully tested the workflow with the following Singularity versions:
-`v2.4.5`
> **NOTE:**
-`v2.6.2`
> The workflow has been tested with the following Singularity versions:
-`v3.5.2`
> * `v2.6.2`
> * `v3.5.2`
After installing Singularity, install the remaining dependencies with:
After installing Singularity, install the remaining dependencies with:
```bash
```bash
conda env create -finstall/environment.yml
conda env create -finstall/environment.yml
```
```
#### As root user on Linux
### As root user on Linux
If you have a Linux machine, as well as root privileges, (e.g., if you plan to
If you have a Linux machine, as well as root privileges, (e.g., if you plan to
run the workflow on your own computer), you can execute the following command
run the workflow on your own computer), you can execute the following command
...
@@ -82,7 +91,7 @@ to include Singularity in the Conda environment:
...
@@ -82,7 +91,7 @@ to include Singularity in the Conda environment:
conda env create -finstall/environment.root.yml
conda env create -finstall/environment.root.yml
```
```
### Activate environment
## 4. Activate environment
Activate the Conda environment with:
Activate the Conda environment with:
...
@@ -90,7 +99,9 @@ Activate the Conda environment with:
...
@@ -90,7 +99,9 @@ Activate the Conda environment with:
conda activate zarp
conda activate zarp
```
```
### Installing non-essential dependencies
# Extra installation steps (optional)
## 5. Non-essential dependencies installation
Most tests have additional dependencies. If you are planning to run tests, you
Most tests have additional dependencies. If you are planning to run tests, you
will need to install these by executing the following command _in your active
will need to install these by executing the following command _in your active
...
@@ -100,38 +111,33 @@ Conda environment_:
...
@@ -100,38 +111,33 @@ Conda environment_:
conda env update -finstall/environment.dev.yml
conda env update -finstall/environment.dev.yml
```
```
## Testing the installation
## 6. Successful installation tests
We have prepared several tests to check the integrity of the workflow and its
We have prepared several tests to check the integrity of the workflow and its
components. These can be found in subdirectories of the `tests/` directory.
components. These can be found in subdirectories of the `tests/` directory.
The most critical of these tests enable you execute the entire workflow on a
The most critical of these tests enable you to execute the entire workflow on a
set of small example input files. Note that for this and other tests to complete
set of small example input files. Note that for this and other tests to complete
@@ -101,27 +101,26 @@ Visual representation of workflow. Automatically prepared with
...
@@ -101,27 +101,26 @@ Visual representation of workflow. Automatically prepared with
Parameter name | Description | Data type(s)
Parameter name | Description | Data type(s)
--- | --- | ---
--- | --- | ---
sample | Descriptive sample name | `str`
sample | Descriptive sample name. <br>**NOTE**: samples split in multiple fastq files (multilane samples), can be automatically merged by using the same ID| `str`
seqmode | Required for various steps of the workflow. One of `pe` (for paired-end libraries) or `se` (for single-end libraries). | `str`
seqmode | There are two allowed values `pe` (paired-end) and `se` (single-end) according to the protocol used. | `str`
fq1 | Path of library file in `.fastq.gz` format (or mate 1 read file for paired-end libraries) | `str`
fq1 | Path of library file in `.fastq.gz` format (or mate 1 read file for paired-end libraries). | `str`
index_size | Required for [STAR](#third-party-software-used). Ideally the maximum read length minus 1 (`max(ReadLength)-1`). Values lower than maximum read length may result in lower mapping accuracy, while higher values may result in longer processing times. | `int`
kmer | Required for [Salmon](#third-party-software-used). Default value of 31 usually works fine for reads of 75 bp or longer. Consider using lower values if poor mapping is observed. | `int`
fq2 | Path of mate 2 read file in `.fastq.gz` format. Value ignored for for single-end libraries. | `str`
fq2 | Path of mate 2 read file in `.fastq.gz` format. Value ignored for for single-end libraries. | `str`
fq1_3p | Required for [Cutadapt](#third-party-software-used). 3' adapter of mate 1. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. | `str`
fq1_3p | Required for [Cutadapt](#third-party-software-used). 3' adapter of mate 1. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. | `str`
fq1_5p | Required for [Cutadapt](#third-party-software-used). 5' adapter of mate 1. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. | `str`
fq1_5p | Required for [Cutadapt](#third-party-software-used). 5' adapter of mate 1. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. | `str`
fq2_3p | Required for [Cutadapt](#third-party-software-used). 3' adapter of mate 2. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_3p | Required for [Cutadapt](#third-party-software-used). 3' adapter of mate 2. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_5p | Required for [Cutadapt](#third-party-software-used). 5' adapter of mate 2. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_5p | Required for [Cutadapt](#third-party-software-used). 5' adapter of mate 2. Use value such as `XXXXXXXXXXXXXXX` if no adapter present or if no trimming is desired. Value ignored for single-end libraries. | `str`
organism | Name or identifier of organism or organism-specific genome resource version. Has to correspond to the naming of provided genome and gene annotation files and directories, like "ORGANISM" in the path below. Example: `GRCh38` | `str`
gtf | Required for [STAR](#third-party-software-used). Path to gene annotation `.fa` file. File needs to be in subdirectory corresponding to `organism` field. Example: `/path/to/GRCh38/gene_annotations.gtf` | `str`
gtf_filtered | Required for [Salmon](#third-party-software-used). Path to filtered gene annotation `.gtf` file. File needs to be in subdirectory corresponding to `organism` field. Example: `/path/to/GRCh38/gene_annotations.filtered.gtf` | `str`
genome | Required for [STAR](#third-party-software-used). Path to genome `.fa` file. File needs to be in subdirectory corresponding to `organism` field. Example: `/path/to/GRCh38/genome.fa` | `str`
sd | Required for [kallisto](#third-party-software-used) and [Salmon](#third-party-software-used), but only for single-end libraries. Estimated standard deviation of fragment length distribution. Can be assessed from, e.g., BioAnalyzer profiles. Value ignored for paired-end libraries. | `int`
mean | Required for [kallisto](#third-party-software-used) and [Salmon](#third-party-software-used), but only for single-end libraries. Estimated mean of fragment length distribution. Can be assessed, e.g., from BioAnalyzer profiles. Value ignored for paired-end libraries. | `int`
libtype | Required for [Salmon](#third-party-software-used), and, after internal conversion, for [kallisto](#third-party-software-used) and [ALFA](#third-party-software-used) . See [Salmon manual][docs-salmon] for allowed values. **WARNING**: do *NOT* use `A` to automatically infer the salmon library type, this will cause kallisto and ALFA to fail. | `str`
fq1_polya3p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 3' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. | `str`
fq1_polya3p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 3' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. | `str`
fq1_polya5p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 5' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. | `str`
fq1_polya5p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 5' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. | `str`
fq2_polya3p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 3' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_polya3p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 3' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_polya5p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 5' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. Value ignored for single-end libraries. | `str`
fq2_polya5p | Required for [Cutadapt](#third-party-software-used). Stretch of `A`s or `T`s, depending on read orientation. Trimmed from the 5' end of the read. Use value such as `XXXXXXXXXXXXXXX` if no poly(A) stretch present or if no trimming is desired. Value ignored for single-end libraries. | `str`
index_size | Required for [STAR](#third-party-software-used). Ideally the maximum read length minus 1. (`max(ReadLength)-1`). Values lower than maximum read length may result in lower mapping accuracy, while higher values may result in longer processing times. | `int`
kmer | Required for [Salmon](#third-party-software-used). Default value of 31 usually works fine for reads of 75 bp or longer. Consider using lower values if poor mapping is observed. | `int`
organism | Name or identifier of organism or organism-specific genome resource version. Has to correspond to the naming of provided genome and gene annotation files and directories, like "ORGANISM" in the path below. <br>**Example:**`GRCh38` | `str`
gtf | Required for [STAR](#third-party-software-used). Path to gene annotation `.gtf` file. File needs to be in subdirectory corresponding to `organism` field. <br>**Example:**`/path/to/GRCh38/gene_annotations.gtf` | `str`
genome | Required for [STAR](#third-party-software-used). Path to genome `.fa` file. File needs to be in subdirectory corresponding to `organism` field. <br>**Example:**`/path/to/GRCh38/genome.fa` | `str`
sd | Required for [kallisto](#third-party-software-used) and [Salmon](#third-party-software-used), but **only** for single-end libraries. Estimated standard deviation of fragment length distribution. Can be assessed from, e.g., BioAnalyzer profiles | `int`
mean | Required for [kallisto](#third-party-software-used) and [Salmon](#third-party-software-used), but **only** for single-end libraries. Estimated mean of fragment length distribution. Can be assessed, e.g., from BioAnalyzer profiles | `int`
libtype | Required for [Salmon](#third-party-software-used), and, after internal conversion, for [kallisto](#third-party-software-used) and [ALFA](#third-party-software-used). See [Salmon manual][docs-salmon] for allowed values. <br>**WARNING**: do *NOT* use `A` to automatically infer the salmon library type, this will cause kallisto and ALFA to fail. | `str`

{kind=link}
{kind=link}