

-
- Downloads
Testing new functions locally
Showing
- .github/.gitignore 1 addition, 0 deletions.github/.gitignore
- .github/ISSUE_TEMPLATE/issue_template.md 16 additions, 0 deletions.github/ISSUE_TEMPLATE/issue_template.md
- .gitignore 6 additions, 0 deletions.gitignore
- DESCRIPTION 6 additions, 3 deletionsDESCRIPTION
- NAMESPACE 4 additions, 0 deletionsNAMESPACE
- R/change_data_format_to_longer.R 32 additions, 12 deletionsR/change_data_format_to_longer.R
- R/data_binding.R 42 additions, 55 deletionsR/data_binding.R
- R/generate_qu_path_script.R 2 additions, 4 deletionsR/generate_qu_path_script.R
- R/get_QC_plots_and_stats.R 164 additions, 0 deletionsR/get_QC_plots_and_stats.R
- R/make_count_dataframe.R 14 additions, 30 deletionsR/make_count_dataframe.R
- R/make_run_config.R 23 additions, 3 deletionsR/make_run_config.R
- R/parsers.R 82 additions, 0 deletionsR/parsers.R
- README.md 77 additions, 31 deletionsREADME.md
- assets/QC_plot1.png 0 additions, 0 deletionsassets/QC_plot1.png
- assets/QC_plot2.png 0 additions, 0 deletionsassets/QC_plot2.png
- assets/QC_plot3.png 0 additions, 0 deletionsassets/QC_plot3.png
- inst/extdata/to_merge/B38_Ascites_GP.csv 968 additions, 968 deletionsinst/extdata/to_merge/B38_Ascites_GP.csv
- man/data_binding.Rd 3 additions, 3 deletionsman/data_binding.Rd
- man/get_QC_plots_parsed_merged_data.Rd 55 additions, 0 deletionsman/get_QC_plots_parsed_merged_data.Rd
- man/make_run_config.Rd 4 additions, 1 deletionman/make_run_config.Rd
.github/.gitignore
0 → 100644
.github/ISSUE_TEMPLATE/issue_template.md
0 → 100644
R/get_QC_plots_and_stats.R
0 → 100644
R/parsers.R
0 → 100644
assets/QC_plot1.png
0 → 100644
{kind=link}
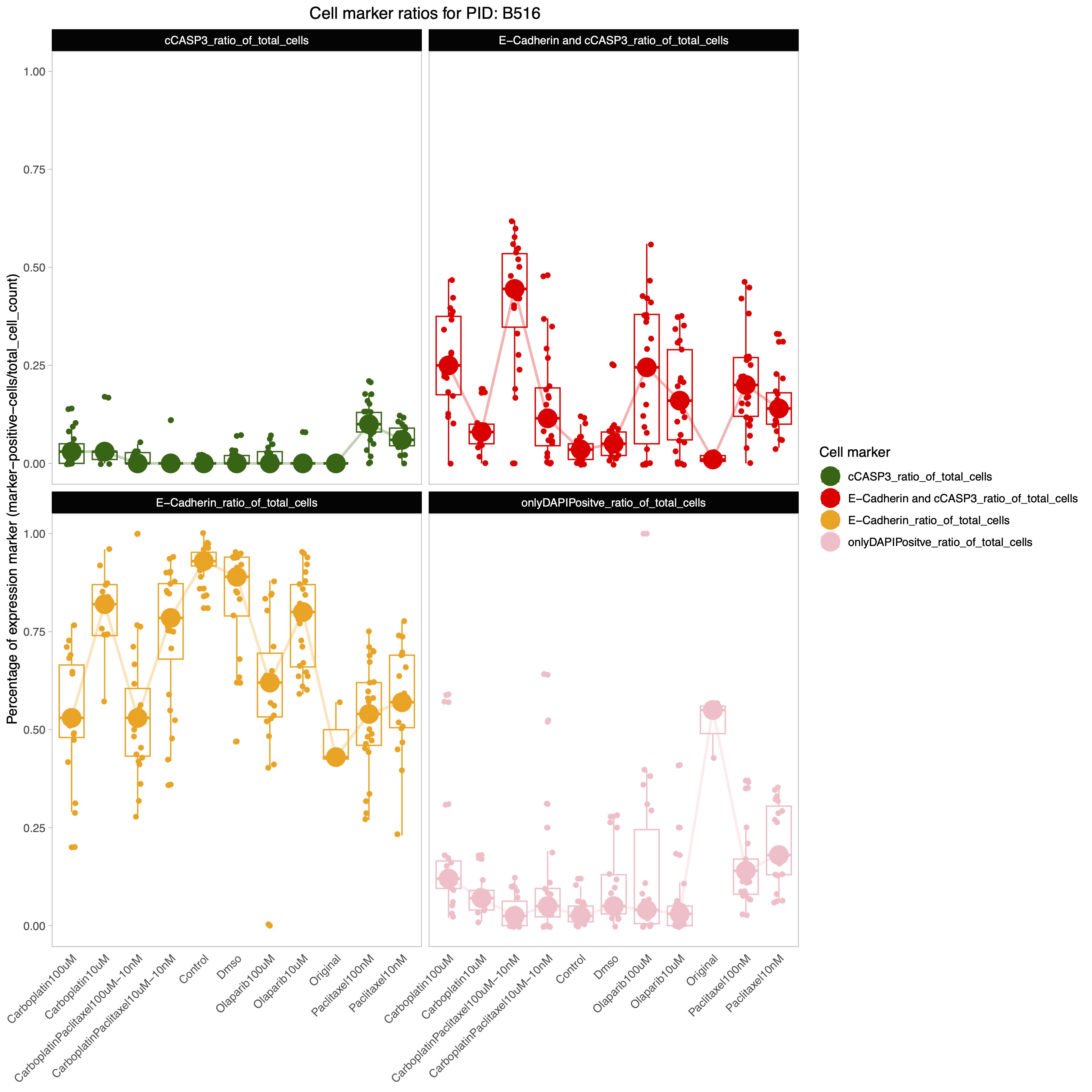
821 KiB
assets/QC_plot2.png
0 → 100644
{kind=link}
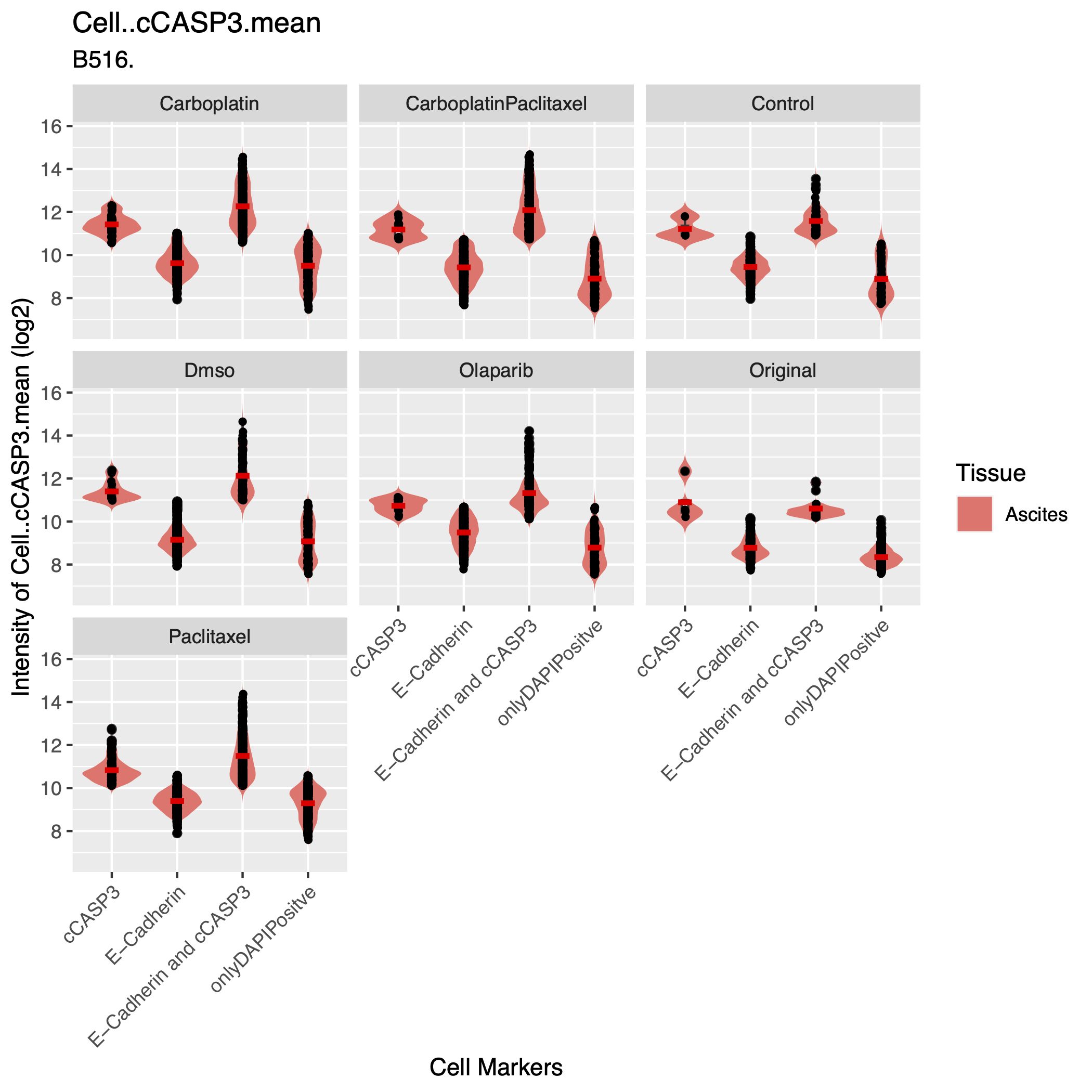
364 KiB
assets/QC_plot3.png
0 → 100644
{kind=link}
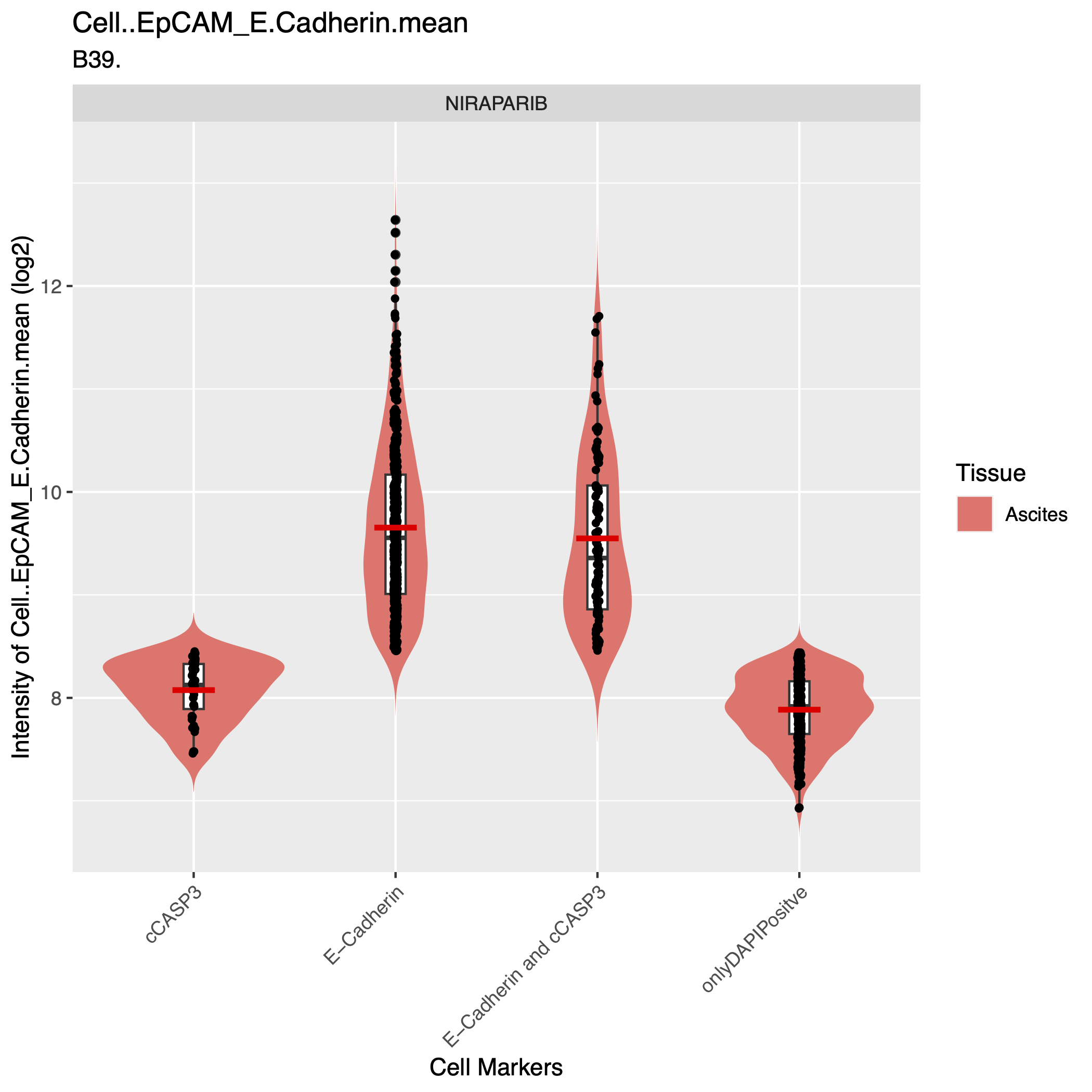
253 KiB
Source diff could not be displayed: it is too large. Options to address this: view the blob.
man/get_QC_plots_parsed_merged_data.Rd
0 → 100644